HEREDITARY HEMORRHAGIC TELANGIECTASIA
NANCY W. BURKHART, BSDH, EdD
Your patient today is a nine-year-old female who is in your practice for an exam and maintenance appointment. Her name is Maggie, and her father accompanies her. As you begin the appointment today, you review the medical history and you learn that Maggie was previously diagnosed with Rendu-Osler-Weber disease (see NIH website on inherited diseases). The disease is also known as Osler-Rendu-Weber syndrome and hereditary hemorrhagic telangiectasia (HHT).
Although the entity is rare, you have actually treated a patient with this disorder, so you feel rather comfortable discussing this with Maggie's father. You know the importance of determining if Maggie is being monitored by a health-care individual for her medical condition.
Rendu-Osler-Weber Syndrome (HHT) is an autosomal dominant blood vessel disorder, which means that one altered gene in each cell will cause the disorder. The most common forms of HHT (types I and II) occur because of a mutation in ENG (encoding the endoglin protein) located on gene 9q33-34 (Type 1) or the activin receptor-like kinase-1 (ALK-1) gene located on chromosome 12q13 (Type 11). These chromosomes produce a protein substance that is involved in blood vessel formation, maintaining endothelial wall integrity, modulating different cell processes, and organization. The arterioles, capillaries, and venules are all affected, causing the vessels to become engorged and dilated.
Epidemiology - Children have a 50% chance of having ROW/HHT if one parent has the disorder. ROW syndrome affects males and females equally. The defective genes are passed from generation to generation, but some individuals have mild forms. Sometimes, it appears to have skipped a generation only to be seen with a later generation. The vascular disorder affects one in 5,000 individuals (Faughnan et al. 2011). This number is skewed since some individuals have mild forms and are never diagnosed.
According to the HHT Foundation International, approximately 1.2 million people worldwide have this disorder, which is uncommon but not rare. The rates of those affected vary greatly with the highest rate being in parts of the Dutch Antilles in the Afro Caribbean with a rate of 1 in 200 persons. In France, the rate is 1 in 8,345 and in England the rate is 1 in 39,216. Over 90% of patients are diagnosed by age 40, but with mild forms, some patients may not be recognized or diagnosed in childhood. They may be diagnosed later in life as the disorder may become more pronounced. For instance, 25% to 30% of individuals develop GI bleeding in their 50s to 60s. Stroke and life-threatening hemorrhage may occur in children or adults and anemia may also be problematic for these patients.
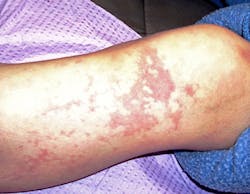
Extraoral characteristics - External characteristics include blood vessel problems such as prominent spiderlike skin lesions and mucocutaneous telangiectases (dilated blood vessels). These characteristics draw attention to the disorder and are especially noticeable on the external skin surfaces (see Figure 1). The visible external and mucosal effects will vary with the extent of the disease process.
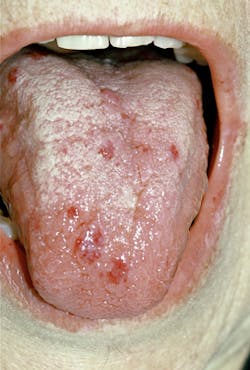
The small "spider veins" will blanch with pressure. The face, lips, mouth, nares, ears, tongue, chest, feet, and visible body areas are most affected and easily viewed by the practitioner. The lips and tongue are prime areas for small, pinpoint-size telangiectases. See Figure 2 of an adult male. Although many people have benign "red spots" from time to time, over 90% of the patients with HHT will usually have multiple areas on a regular basis.
Internal characteristics - Internal organs such as the brain, liver, lungs, spleen, urinary tract, and spine may be affected with arteriovenous malformations (AVM) that can be life-threatening and may go undiagnosed. Over 30 to 35% of patients with HHT develop AVM in their lungs (HHT Foundation International). Epistaxis (bleeding from the nose) is often a key factor in the disorder and especially relevant in those with a family history of ROW, and this occurs in over 90% of patients. The nosebleeds are the most common problem and humidifiers are suggested to keep the tissues moist. Laser therapy is often used for skin/nasal treatment and patients see improvement for longer periods of time.
Life-threatening events related to AVM of the brain, lungs, stomach, intestines, and liver are more serious. Treatment will depend upon the extent of the dilated vessels and the numerous problems that may occur. According to the HHT Foundation International, approximately 25% of patients will develop gastrointestinal bleeding, usually in later life.
Diagnosis - Most patients are diagnosed by the Curacao criteria established in 1999 by the scientific advisory board of the HHT Foundation International. The diagnosis of HHT is made both clinically and through genetic testing. When three or more of the following occur, a definitive diagnosis is usually made (from the HHT Foundation International):
• Nosebleeds - spontaneous and recurrent
• Telangiectasia - Multiple, at characteristic sites, including lips and oral cavity, fingers, and nose
• Internal telangiectases or AVM, lung, brain, GI, liver, or spinal
• First-degree family member such as a parent, sibling, or child diagnosed with HHT.
The use of MRI, oxygen saturation, echocardiogram, CT scan along with other tests assists in providing a definitive diagnosis of the current state of the patient's health. They are also used periodically in checking the progression of the disease (Sekarski & Spangenberg, 2011).
Patients who have any of the clinical signs or just a family history of HHT should be followed, tested, and monitored since some manifestations may be missed with internal organs even though the person may have no physical signs, such as skin telangiectases or epistaxis. A thorough medical history, family history, and documentation are necessary. A referral for evaluation is needed to a center or a physician who specializes in the disorder at a treatment facility. Other family members should be screened as well.
Dental implications - The dental professional is key to advising these patients to seek care in order to avoid future problems especially when a definitive diagnosis and treatment may not have occurred. The oral cavity and especially the tongue are prime areas for telangiectases. Any "red spots" noted in the oral area should be questioned and an etiology needs to be determined.
Patients may have chronic oral ulcers and vesicles as well as bleeding when brushing because of diminished vascular wall thickness associated with inflammation (Hopp et al. 2012). Gingival bleeding upon scaling should be a prime concern. The treatment goal is to manage and prevent hemorrhage.
NSAIDS and aspirin should be avoided in patients with HHT because of the prolonged bleeding that may occur, especially with nosebleeds. American Heart Association guidelines should be closely followed with regard to subacute bacterial endocarditis (SBE).
Dentists and hygienists are of paramount importance in early detection of not only oral cancer, but also other disease states that have clear medical implications. Patients may see a dental professional yearly and may only see a medical professional for a specific problem.
Many of the signs related to HHT such as telangiectasia are visible on external skin surfaces and may give the practitioner a clue before even completing the intraoral exam. Young patients may not have been diagnosed and need referral for a definitive diagnosis to a specialist. The earlier the diagnosis is made, the more favorable the long-term management of HHT.
Remember to keep asking good questions, and always listen to your patients.
REFERENCES
Buckmiller LM, Richter GT, Suen JY. Diagnosis and management of hemangiomas and vascular malformations of the head and neck. Oral Dis. 2010; 16(5): 405-418.
Hopp RN, Cardoso de Siqueira DC, Sena-Filho M, Jorge J. Oral vascular malformation in a patient with hereditary hemorrhagic telangiectasia: a case report. Spec Care Dentist. 2012; 32(1): 11-14.
Osler W. On a family form of recurring epistaxis associated with multiple telangiectases of the skin and mucous membranes. Bull Johns Hopkins Hosp: 1901;12:333-337.
Sekarski LA, Spangenberg LA. Hereditary Hemorrhagic Telangiectasia. Pediatr Nurs. 2011; 37(4):163-168.
Faughnan ME, Palda VA, Garcia-Tsao G, Geisthoff UW, McDonald J, et al. HHT Foundation International - Guidelines Working Group.
J Med Genet. 2011 Feb;48(2):73-87. Epub 2009 Jun 23.
Westerman CJ, Rosina AF, De Vries V, et al. The prevalence and manifestations of hereditary hemorrhagic telangiectasia in the Afro-Caribbean population of the Netherlands Antilles: a family screening. Am J Med Genet A. Feb 1 2003; 116A(4):324-8.
Web sites:
HHT Foundation International
hht.org/about-hht/
hht.org/about-hht/facts-at-a-glance/
Vascular Disease Foundation
http://www.vdf.org
National Center for Advancing Translational Sciences Office of Rare Diseases Research
http://www.vdf.org